Frequently Asked Questions about Biosimilar Medicines
"Healthcare biotech is a fast growing field within modern medicine. It can play a vital role in tailoring healthcare to the individual as well as providing treatments for hitherto incurable diseases."
Biological and Biosimilar Medicines
Biotechnology has enabled us to find cures for some of the most serious diseases known to man. Worldwide, the lives of over 300 million people have been transformed by the availability of a growing number of biotechnology drugs. Today patients have access to more than 150 biotechnology drugs and vaccines, and medical science is working to increase that number every day. But biotechnology is a complicated science. As the patents of certain biological medicines are expiring, follow on versions are being developed. This fact sheet takes a look below the surface and explains the complexities and different properties of biological medicines or 'biologics' and the consequences for so-called follow-on biologics or biosimilar medicines.
WHY are biosimilar medicines being introduced?
Medicinal products developed through biotechnology constitute an essential part of medicines available to patients today. They represent approximately 6% of the pharmaceuticals currently marketed and account for more than 9% of total pharmaceutical expenditure. More importantly, 'a third of products in the development pipeline are biotechnology products. Oncology is the largest area.'
Some major biotechnology-derived medicines are, or will soon, no longer be protected by patents. As for all other medicines when their 20-year patent expires, they will become open to development and manufacture by other companies. This introduces competition on the market which ensures patient access to safe and effective — and more affordable —biotechnology-derived medicines. Without competition the prices of the originator biotechnology-derived medicines would remain artificially high. Similarly, this competition will serve to stimulate research into new originator medicines. This fact is borne out by the situation in the USA where more than 50% of medicines used are generic medicines and where, at the same time, more new originator medicines are developed than anywhere else in the world.
In addition, Europe has a critical need to control healthcare costs. Europe cannot afford not to have competition from biosimilar products. There were already more than 72 million people aged 60 and over in the EU-15 alone at the end of 2005 and that number is expected to grow by 25% in those countries by 2015. This will equate to an additional 50 million people over age 60 and these numbers will, of course, be even higher for the full EU-27. Since this age group spends on average three to four times more on medicines than when they were 30, the cost of providing adequate access to medicines for these people is exploding. Many biopharmaceuticals are also often used to treat long-term conditions such as diabetes, cancer, chronic kidney failure and multiple sclerosis. On average, biopharmaceuticals cost much more per patient than conventional pharmaceuticals, and their use is growing at more than 20% per year. As already noted, the biologicals pipeline is expanding. Study after study shows that not all patients who could benefit from these medicines have access to them — and with pharmaceutical spending growing more than twice as fast as the gross domestic product (GDP), that situation can only worsen. It is therefore critical that everything possible be done to maximise patient access to cost effective biopharmaceuticals — and this means rapid introduction of biosimilar medicines as soon as patents expire.
Another important reason for introducing biosimilar medicines is, of course, that the scientific basis for biosimilar development and technology exists to obtain approval and bring this new type of medicines to the market. As a result, a legal framework has been established in Europe to govern their development and approval.
WHERE is Europe now regarding biosimilar medicines?
Biosimilar medicines are now a reality in the European Union. The necessary legal framework for biosimilar medicines has been solidly established in the EU and the first biosimilar medicines were approved by the European Commission in April 2006, with the endorsement that each of them "has been compared to and matches the reference medicine [...] in terms of quality (how it is made), safety (for example the side effects that can occur when receiving treatment are similar), and effectiveness." Guidance on risk management systems has also been developed which assures safe market entry and post-marketing monitoring of these medicines.
In addition, a legal framework has been in place since 30 October 2005, which allows the performance of tests and trials in view of obtaining authorisation for a biosimilar medicine in the European Union without breaching patent law. This will facilitate the development of biosimilar medicines on the European territory.
WHAT is the regulatory approval process for biosimilar medicines in Europe?
All biotechnology medicines, including biosimilar biotechnology-derived medicines, are or will be assessed by the European Medicines Agency in London (EMEA), which constitutes the scientific body of the European Commission responsible for the evaluation of medicines. They are approved by the European Commission based on the positive scientific opinion issued by the EMEA.
When the EMEA assesses data for a biosimilar medicine, the scientific principles for ensuring product quality, safety and efficacy are identical to those applied to the originator/brand reference medicine with which comparability is demonstrated
In addition to the quality data required for all biotechnology products, the companies involved in the developing biosimilar medicines must additionally submit 'comparability data'. Indeed, manufacturers must characterise, in parallel, both their biosimilar product and the originator reference product. They must demonstrate, with a high degree of certainty, that the quality of the biosimilar medicine is comparable to the originator/reference medicinal product. A comparability programme is clearly defined and agreed upon in advance with the EMEA, who defines the set of non-clinical and clinical data that are necessary to sufficiently demonstrate biosimilarity. The extent of this data varies according to the type and complexity of the medicine involved. Each individual biosimilar medicine is assessed on a case-by-case basis.
HOW can patients be assured of quality?
Both originator reference products and biosimilar medicines are made under carefully controlled conditions to ensure the products are consistent and of the required quality. This is known as Good Manufacturing Practice (GMP). In the European Union, GMP inspections for all biopharmaceuticals — originator reference products and biosimilar medicines — are coordinated by the EMEA and performed by the national regulatory agencies. There are three levels:
Routine GMP inspections of manufacturing sites to ensure that medicines are produced safely and correctly.
Pre-Approval Inspections (PAI) for GMP compliance prior to the approval of new medicines for marketing if there are specific issues identified during the approval process.
Unannounced inspections.
HOW can patients be assured of safety?
All European pharmaceutical companies are legally required to monitor the use and effects of all their medicines. They must have systems in place to detect, assess, understand and endeavour to communicate any adverse reactions or any other medicine-related problem. The science and activities of these processes are known as 'Pharmacovigilance'. As part of the pharmacovigilance process each manufacturer must provide a description of their pharmacovigilance system before its medicine is approved. This system is inspected by the regulatory authorities.
In addition, for every new medicine, including biosimilar medicines, a Risk Management Plan (RMP) must be submitted and agreed to by the EMEA. The RMP describes what is known about the safety of the medicine and outlines how the manufacturer will further monitor and fill any gaps in knowledge as well as any measures needed to minimise any risk from the medicine. This plan must be regularly updated throughout the entire time the medicine is marketed and used.
Once marketed, pharmaceutical companies must also continuously evaluate the information on the benefits and risks of their medicines.
WHAT measures are needed to ensure access to biosimilar medicines?
Strategies to increase the affordability of medicines — and to make them available to more patients — include promoting competition and generic medicines, including generic substitution. Biosimilar medicines need to be included in these strategies. Now that a solid legal and regulatory environment has been established, a clear market pathway for biosimilar medicines needs further consideration in each individual Member State to enable access to these medicines as soon as possible after their marketing approval. Indeed, the way countries determine which medicines are selected and used needs to be adjusted to ensure biosimilar medicines are made available. Factors include marketing authorisation approval, agreement on price and on how the biosimilar medicines are reimbursed, and gaining the acceptance of clinicians and patients to using biosimilar medicines as part of their practice and treatment. Access for patients to biosimilar medicines is not automatic; it requires that proactive steps be taken by all relevant stakeholders.
WHAT cost savings will biosimilar medicines bring to healthcare systems?
The improved affordability of healthcare that could result from the use of biosimilar medicines is real. It has been estimated that a 20% reduction on six off-patent, or soon-to-be off-patent, biopharmaceuticals would save the EU over €1.6 billion.
The price differential between a reference product and a biosimilar medicine will depend on the relative development costs. Biosimilar medicines can be expected to be offered at a price below that of the reference product, partly as a result of production process efficiencies, and partly because of the reduced costs of a streamlined development programme. The greatest savings are likely to result from the clinical trial programme, since a biosimilar medicine (containing a well-known and well-used substance) would require less clinical data to support its approval. This price differential should lead to a significant and much-needed release of healthcare funds.
WHY should patients be involved in the biosimilars debate?
Patients need to contribute their views actively to this debate to ensure that cost effective biotechnology-derived therapies are made available to them as quickly as possible. Patients should discuss access, cost, safety and efficacy of receiving biosimilar medicines as these are now an emerging source of affordable medicines for more patients with some of the most difficult to treat diseases.
Information about specific biosimilar medicines is available from a number of sources, including the EMEA, to enable patients, together with their doctor and pharmacist, to have access to the relevant data on the use of these medicines.
Patients can have confidence in biosimilar medicines because they are approved by the same regulatory organisation, with the same scientific rigour, and using the same regulatory systems as the comparable originator products.
Healthcare teams also need to live up to their responsibility to contribute to patients' awareness of the existence of biosimilar medicines and to communicate their confidence in the scientific assessment performed by the EU regulatory authorities.
HOW is the landscape evolving for biosimilar medicines?
By 2008-2012 will see further developments in relation to biosimilar medicines, and healthcare professionals and healthcare purchasers need to ensure that they are aware of what is happening in this rapidly changing environment.
The patent protection on a large number of originator reference products has expired since 2001, which would lead to expect a growing number of biosimilar products on the market in the not too distant future.
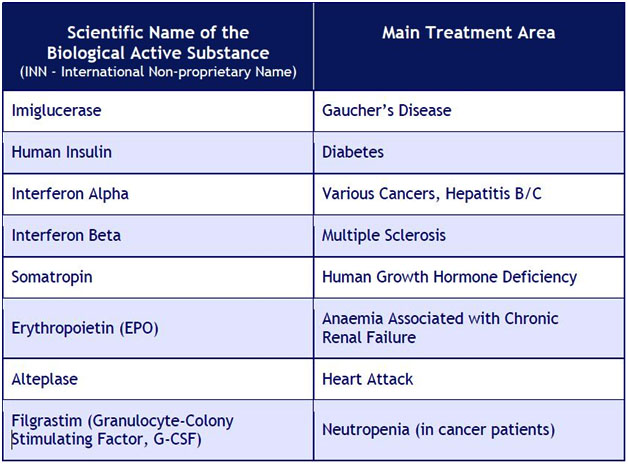
EXAMPLE ACTIVE SUBSTANCES of originator reference products with their main areas of treatment
Content Disclaimer
Contents in our website are from external websites. Links to and content from external website are provided for the convenience of users. Biopractice.com takes no responsibility for the content of such links and websites.